LLC et traitement précoce à l’ère des thérapies ciblées
Ibrutinib versus placebo pour les patients atteints de LLC de faible risque, résultats de l’essai de phase III randomisé en double aveugle CLL12.
Ibrutinib versus placebo in patients with asymptomatic, treatment-naïve early stage chronic lymphocytic leukemia (cll): final results of the phase 3, double-blind, placebo-controlled cll12 trial.
D’après la communication orale de Petra Langerbeins et al. Abstract #S200, EHA 2023.
Contexte de l’étude
Les patients atteints de LLC de stade limité (Binet A, RAI 0-II) n’ont pas de symptômes, et les recommandations actuelles ne sont pas en faveur de l’initiation d’un traitement (NCCN, iwCLL). Cette recommandation est principalement basée sur des essais d’immunochimiothérapie s’intéressant à ce groupe de patients et qui n’ont pas réussi à démontrer un bénéfice en survie par rapport à l’attente du développement d’une maladie symptomatique pour débuter. La même question de l’intérêt d’initier un traitement précocement avant tout signe de pathologie se pose désormais dans l’ère des thérapies ciblées avec l’ibrutinib.
Objectifs de l’étude
Les patients à ‘haut risque’ de progression sont définis par le score pronostic du groupe allemand de la LLC à partir de critères d’âge (>60ans), de sexe (masculin), de taux de béta-microglobuline, de PS (>0), d’activité de thymidine-kinase (>10), ou par la présence d’IgHV non mutées, la présence de del11q ou de del17p. Les patients ont été classés en groupe « sans risque », et simplement suivis (non analysés), tandis que les patients à risque augmenté étaient randomisés entre ibrutinib et placebo, en double aveugle. Pour ce qui est du plan d’analyse statistique, il fallait bien connaître CLL12 (et sa publication), en effet à aucun moment des détails n’ont été donnés sur les hypothèses statistiques dans la présentation. L’objectif est la démonstration d’une supériorité en termes de survie sans évènement (SSE) dans le groupe ibrutinib. Les évènements considérés sont le temps depuis la randomisation jusqu’à la survenue d’une indication de traitement, ou d’une initiation ou du décès. Les analyses ont été menées en intention de traiter. À noter que la survie globale, qui faisait partie des analyses secondaires n’a pas pu être analysée, par manque d’évènements.
Résultats de l’étude
L’étude s’est terminée en juillet 2022 après avoir débuté en 2014. Les patients sans risques étaient au nombre de 152, tandis que 363 patients de haut risque ont été randomisés entre 182 Ibru et 181 placebo. Le suivi médian est de 69 mois. Les caractéristiques de la pathologie mettaient en évidence des del17p dans 3-4% des cas, 39% d’IGHV non-mutées, 76-83% d’activité thymidine kinase augmentée et 8% de ß2-microglobuline augmentée.
Paradoxalement les résultats les plus intéressants sont ceux de tolérance/toxicité, puisque 99% des patients ont eu des évènements indésirables (dans les deux bras) et 66-72% des évènements graves, de grade 3-5 (et 15% de SAE dans le groupe placebo contre 10% dans le groupe ibrutinib). Bien entendu, aucun AE dans le groupe « faible risque » qui n’avait pas été randomisé. La différence se creuse lorsque l’on s’intéresse aux effets indésirables d’intérêt clinique : 80% versus 52%, dont 36% versus 15% de saignements, 22% versus 10% d’arythmies, 19% versus 8% d’HTA, 41% versus 29% de diarrhées. On a observé des taux d’incidence élevés de cancers secondaires, 13% dans le groupe ibrutinib, 21% dans le groupe placebo, et 10% dans le groupe « faible risque ».
Le groupe Ibru a reçu en médiane 39 cycles, avec 62% d’arrêt prématuré, tandis que le groupe placebo a reçu 27 cycles et a stoppé précocement dans 81% des cas. Les raisons d’arrêt dans le groupe placebo ont été la progression (48%) et les toxicités (!) pour 29%. Dans le groupe ibrutinib, la progression n’a compté que pour 5% des arrêts, contre 72% pour la toxicité. Dans le bras ibrutinib,
il est à noter que seuls 3 patients ont obtenu une RC et 71% ont obtenu une réponse partielle (on note 5% de RP pour le bras placebo).
Concernant l’objectif principal, l’essai confirme la supériorité du bras Ibru versus placebo avec une SSE non atteinte versus 52 mois (HR=0.28, IC95%=0.2-0.41), figure 1. De la même façon, le temps jusqu’au prochain traitement était en faveur du bras ibrutinib (HR=0.24, IC95%=0.16-0.38).
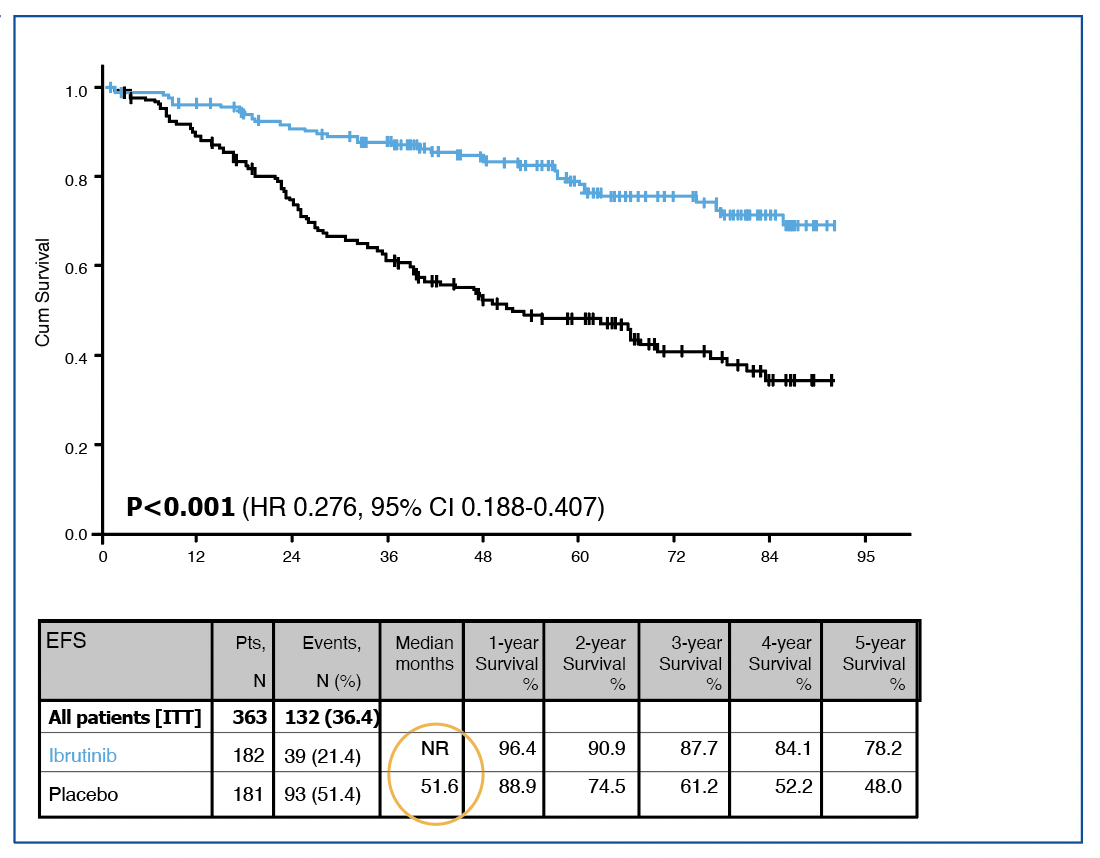
Figure 1 : SSE de l’étude CLL12.
En termes de traitement à la progression, il s’agit d’un 50/50 : la moitié des patients dans les deux groupes ont reçu une immuno-chimiothérapie et l’autre moitié des traitements ciblés, tandis que les patients de faible risque ont reçu à 87% un traitement ciblé.
En ce qui concerne la PFS2 (qui n’en est pas vraiment une puisqu’elle commence après le début du traitement N+1 et pas à la randomisation), l’analyse est en défaveur de l’ibrutinib avec une médiane de PFS2 à 16,6 mois versus 35 mois (HR=1.47, IC95%=0.8-2.6).
Les survies globales sont excellentes (~93% à 5 ans), bien entendu sans différence (HR=0.8). En ce qui concerne les causes de décès (~13 dans chaque bras), il est impossible d’esquisser un message clair tant ils ont été rares (notons tout de même 2 hémorragies intracrâniennes dans le bras ibrutinib). Enfin, lorsqu’on compare les survies globales depuis le diagnostic, les patients randomisés de « haut risque de progression » et les patients de bas risques ont des survies tout à fait superposables, figure 2.
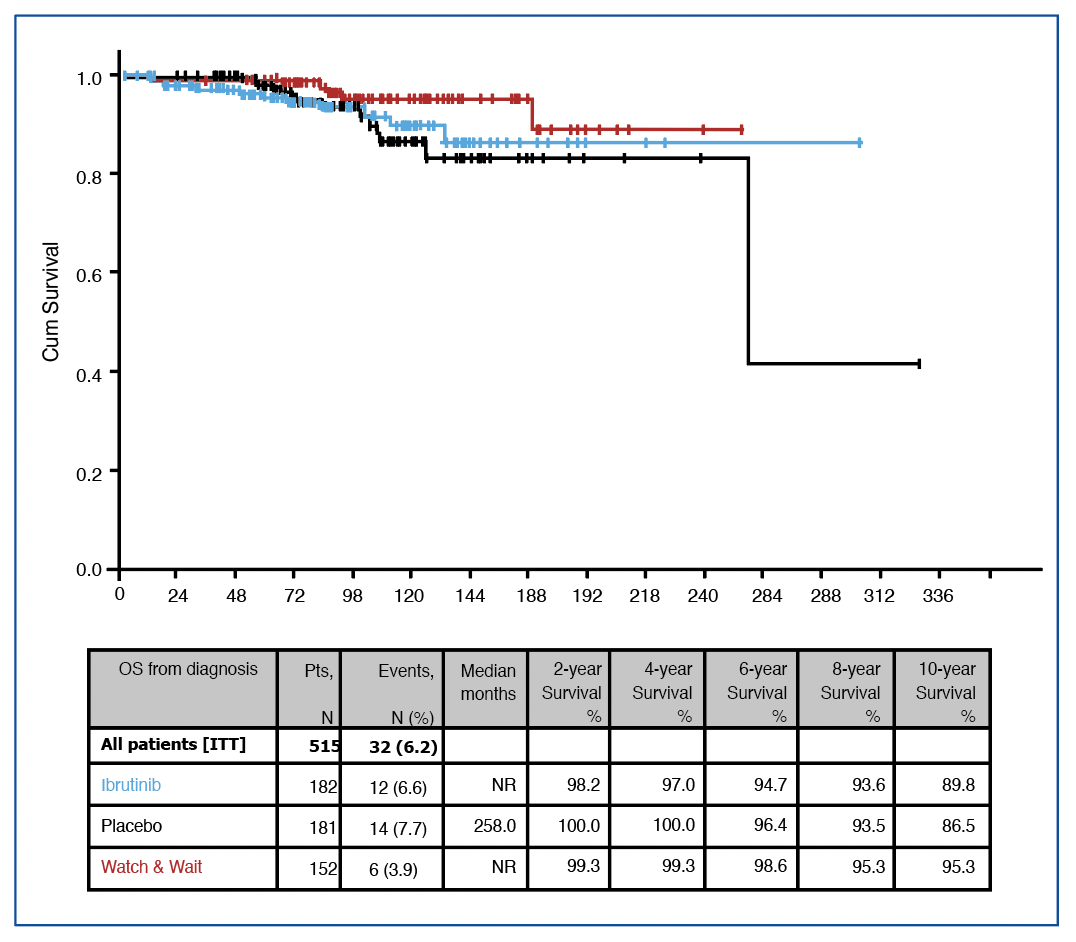
Figure 2 :
survie globale de l’étude CLL12.
Quels impacts sur les connaissances et les pratiques cliniques ?
L’ibrutinib chez ces patients asymptomatiques à haut risque de progression :
- est associé à des toxicités significatives ;
- améliore les critères composites (EFS, PFS, TTNT)…. ;
- … sans améliorer la vie des patients ou leur survie ;
- la stratégie de surveillance doit rester la norme.
Plusieurs essais préemptifs sont en cours dans cette population, en particulier avec le vénétoclax, ou des BTKi de secondaire génération, et ont tous un point en commun : des difficultés importantes de recrutement, on peut saluer le groupe allemand pour avoir fini le travail.
Critique méthodologique
Cette phase 3 suit le gold standard des essais cliniques : randomisé en double-aveugle versus placebo, ce qui donne une indication sur la robustesse des résultats qui viendront. Trois cent soixante-trois patients avec risques accrus ont été inclus, la moitié a reçu de l’ibrutinib, plus un autre bras de 152 patients sans risque pour cette maladie (un groupe nommé « watch and wait »), ce qui n’est pas courant. L’endpoint principal était la survie sans événements (EFS), sans pour autant spécifier quelles étaient les hypothèses de départ (différences de probabilités entre Ibru et placebo, à quel temps depuis la randomisation, puissance…) et le type d’événements à considérer pour cet événement composite (on le saura plus tard dans la présentation) pour le calcul du nombre de patients. La durée médiane de suivi était supérieure à 5 ans.
L’une des premières informations données concerne les événements indésirables (EI), en proportion plus importantes dans le bras ibrutinib (80% contre 52% chez placebo, évidemment la différence est très significative), et la majorité de ces adverses events pour toxicité cardiovasculaire ont conduit à plus d’arrêt d’ibrutinib que de placebo. En revanche, il y a eu bien plus de progressions dans le bras placebo (48,5% contre moins de 5% pour Ibru). Pour rappel, le taux de réponse globale (ORR) dans le bras ibrutinib, très majoritairement des réponses partielles, écrasait celui du bras placebo (72% versus 5% respectivement). La SSE (ou EFS en anglais) est bien supérieure dans le bras ibrutinib avec un risque presque divisé par quatre (HR = 0.276 [0.188-0.407]) et une survie médiane non atteinte avec un suivi maximum à 90 mois. Des informations sont données pour la SSE qui comprend comme événements l’évolution symptomatique de la maladie et/ou une contre-indication du traitement (pour EI ?).
Les analyses pour le time to next treatment et survie sans progression vont dans le même sens également, avec pour cette dernière un HR = 0.17 (ce qui est assez spectaculaire comme résultats).
Pour aller plus loin, les auteurs ont utilisé une ‘PFS2’ qui concerne les patients ayant pris un autre traitement, analysée depuis le début de la prise de cet autre traitement (toujours en comparant les deux bras de l’essai) ce qui ne correspond pas tout à fait à la définition de cet outcome (voir focus statistique (La PFS”2″)). Le groupe de patient « watch and wait » (de meilleur pronostic) vient se greffer dans les analyses de survie globales depuis le diagnostic, où aucune différence statistique n’est décelée entre ce groupe et les 2 bras.
Pour le résumé, on aurait souhaité un regard plus tranché sur la balance bénéfique/risque. En effet, quand on s’aperçoit que, malgré les bénéfices indéniables en termes de SSE et SSP, la survie globale n’est pas si différente entre les trois groupes, cela pose question. Ne faudrait-il pas une réflexion plus approfondie sur les critères combinés d’analyses dans les essais cliniques permettant de mieux évaluer cela ?